
CASES OF THE MONTH
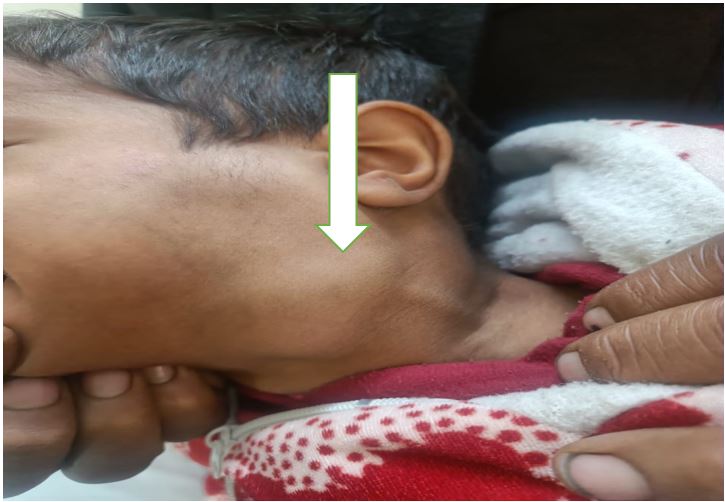
Case :- A 3 year old male presented with massive bilateral cervical lymphadenopathy for 1 year with orbital swelling. Radiology (CECT head & neck) showed multiple homogenous enhancing nodes (largest lymph node measures 23x20mm) and orbital mass.
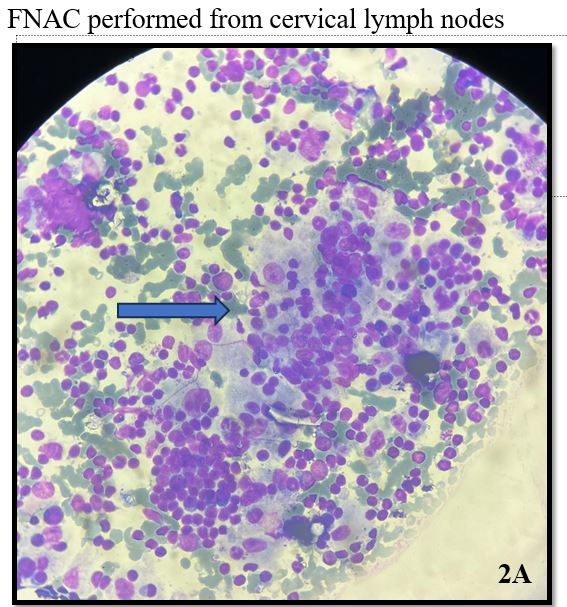
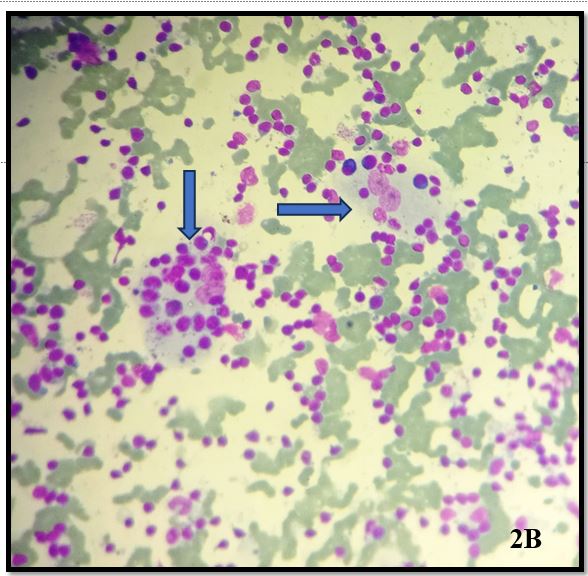
Smears examined show moderate cellularity with prominent population of histiocytes which are characterized by large, round nuclei and voluminous cytoplasm showing emperipolesis with engulfment of lymphocytes, plasma cells and occasionally neutrophils.
No evidence of atypica/mitosis/nuclear grooving/ multinucleated giant cells seen in the smears examined.
—-Rosai Dorfmann Disease
Rosai Dorfman disease (RDD)
- It is a rare, non-Langerhans cell histiocytosis which was initially described as a self limiting condition involving lymph nodes Also known as sinus histiocytosis with massive lymphadenopathy.
- It is a disorder which is characterized by nodal or extranodal accumulation of large, histiocytes/macrophages (S100-positive) which exhibit emperipolesis.
- It can be classic (nodal) or extranodal. Skin is the most common extra-nodal site.
- Classic RDD- common presentation is enlarged lymph nodes (mostly bilateral cervical) in young males.
- Extranodal RDD has varied presentation like cutaneous lesions, neurological, orbital, sinus, pulmonary involvement etc
- Multisystem involvement seen in 20 % cases. Despite its alarming presentation, it is self-limited and simple excision is curative.
- It is associated with other conditions like autoimmune disorders, neoplasia such as lymphomas and myeloid neoplasms and IgG4 related RDD.
- The histiocytes show immunopositivity for monocyte/macrophage marker, S-100, cyclin D1/BCL-1 while negative for langerin, CD1a and BRAFV600E.
- Pathogenesis is unclear and likely multifactorial. Around 50% of cases carry gain-of-function mutations in the MAPK/ERK pathway.
- Its diagnosis involves clinical history, examination, imaging alongwith morphologic features.
- It has a favourable outcome in nodal and cutaneous lesions however, systemic therapies are recommended in disseminated RDD due to its aggressive clinical course.
- On histopathology- the involved lymph node is enlarged with extensive sinusoidal expansion. Nodal architecture is effaced with a diffuse infiltrate of histiocytes. The cortex comprises numerous activated B cells and mature plasma cells with few follicles with pale histiocytes, giving the appearance of alternating dark and light zones. The sinusoids contain numerous large histiocytic cells with smooth contoured hypochromatic nuclei, small distinct centrally placed round nucleoli and ill-defined, pale, wispy cytoplasm. Emperipolesis, a useful but not specific feature, is seen as intact haematolymphoid cells within a vacuole or floating freely in the cytoplasm of the histiocytes.
- Differential diagnosis
• Sinus histiocytosis
• Langerhan’s cell histiocytosis
• Erdheim Chester disease
• Rhinoscleroma
REFERENCES
1.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969 Jan;87(1):63-70.
2.Rajyalakshmi R, Akhtar M, Swathi Y, Chakravarthi R, Bhaskara Reddy J, Beulah Priscilla M. Cytological Diagnosis of Rosai-Dorfman Disease: A Study of Twelve Cases with Emphasis on Diagnostic Challenges. J Cytol. 2020 Jan-Mar;37(1):46-52
3.Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours. Leukemia. 2022;36(7):660-679.
4.Garces S, Medeiros LJ, Patel KP, Li S, Pina-Oviedo S, Li J, Garces JC, Khoury JD, Yin CC. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod Pathol. 2017 Oct;30(10):1367-1377.
5.Goyal G, Ravindran A, Young JR, Shah MV, Bennani NN, Patnaik MM, Nowakowski GS, Thanarajasingam G, Habermann TM, Vassallo R, Sher T, Parikh SA, Rech KL, Go RS; Mayo Clinic Histiocytosis Working Group. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020 Jan 31;105(2):348-357.
- Case 2 :- 6-month-old male child who presented with an abdominal mass and hematuria for 2 months in th surgical OPD.CT abdomen suggested Neoplastic lesion of the kidney.-Rhabdoid tumor Kidney
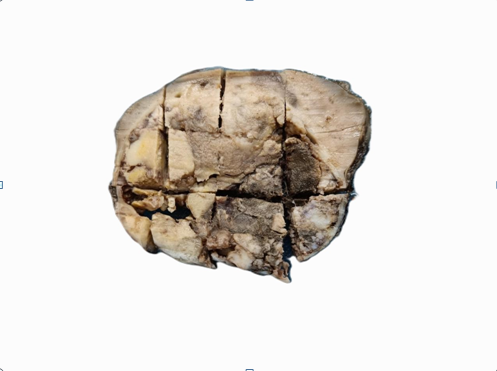
Gross specimen show grey black variegated renal mass destroying the renal parenchyma
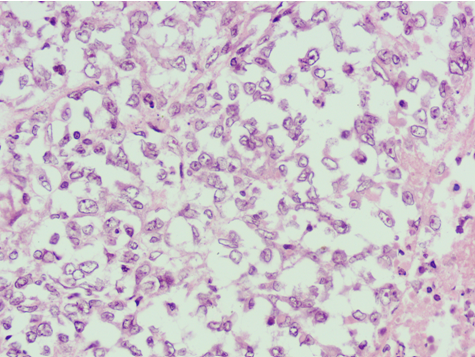
H&E stained sections show atypical rhabdoid cells on 40X
DISCUSSION
The most aggressive renal tumor of young age.
- Highly malignant undifferentiated tumor composed of noncohesive cells with large, eccentric nuclei, prominent nucleoli and intracytoplasmic inclusions; characterized by loss of INI1 immunostaining and SMARCB1genetic abnormalities (in ~95% of cases).
- The majority (~95%) of rhabdoid tumors of kidney have genetic changes resulting in the loss of function of SMARCB1, a tumor suppressor gene encoding BAF47 (INI1), which is a core member of the BAF chromatin remodeling complex
- No established standard treatment.
- Most patients are treated with intensive multimodal regimens; surgical resection, intensive multidrug chemotherapy regimens and local radiotherapy or high dose chemotherapy followed by autologous stem cell rescue.